시판 전 신고 (Premarket Notification또는 510(k))
거의 모든 Class I 제품들은 면제이나 몇몇의 특정한 제품은 시판전 신고(Premarket Notification, 510(k))가 적용 되며,
대부분의 Class II 제품들은 시판전 신고가 요구된다.
510(k)가 필요한 제품들은 미국에 현재까지 유통되는 기존 제품과 상응 또는 유사한 기능(Predicate Device)이라는 것을 증명 해야 한다.
510(k) 제출시 필요한 심사비가 있으며, 심사기간은 대부분이 3개월에서 6개월이지만 때로는 그 이상 소요되기도 한다.
시판 전 승인 (Premarket Approval: PMA)
시판전 승인(Premarket Approval, PMA)이 필요한 것들은 Class III 로 분류된 제품으로서 생명과 건강에 많은 Risk가 따르는 의료기기에 적용된다.
PMA절차에서는 그 제품의 효능을 증명할수 있는 자료와 임상실험이 요구된다.
그러나 일부 Class III 기기는 시판 전 승인이 면제되며 510(k)로 대처될 수 있으므로 미리 철저한 조사가 필요하다. 심사비가 있으며, 심사기간은 약 1년 이상이 소요된다.
510(k)등록을 위한 기본적인 준비문서
- 1. 제품명 및 모델명
- 2. 제품 Label 및 유사제품(predicate device) Label
- 3. 제품설명서(도면, 회로도, 사진등)
- 4. 임상적인 평가 자료 또는 Clinical data
- 5. biocompatibility and test report
- 6. 사용설명서 또는 사용 매뉴얼
- 7. 유사제품(predicate device)과 comparition table
- 8. Manufacture & QC Procedure
- 9. Hazard Assessment
- 10. test report (EMC, Safety, Biological)
- 11. Sterilization validation report
- 12. Software Validation Report
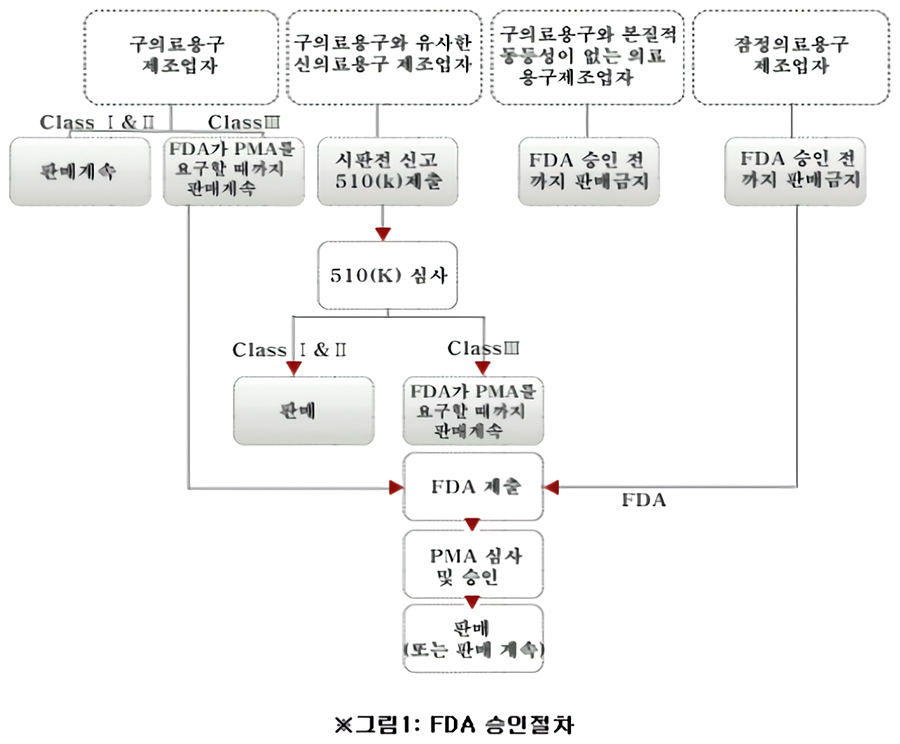
FDA 승인절차
510(k)
미국 FDA에서 Class I(일부 제품) 과 Class II 제품중 이미 허가된 제품과 동등성을 비교를 통하여 제품의 안전성과 유효성을 입증하는 절차입니다.
21 CFR 807.92(a)(3). 조항에 510(k) 에 대한 정의가 나와있습니다.
A 510(k) is a premarket Notification made to FDA to demonstrate that the device to be marketed is at least as safe and effective,
that is, substantially equivalent, to a legally marketed device.
그리고 510(k)를 진행 할때 유사제품과의 비교하여 얼마나 많은 부분이 동등하고 다른부분이 무엇인지가 가장 중요한 요소입니다. 만일 성능적으로 다른 부분이 있다면 이부분에 대한 입증자료를 제출하여야 510(k)가 가능합니다.
Registration
일반적으로 Class I이나 Class II에 해당되는 제품중 제품에 대한 위험성이 작거나 이미 오래전부터 사용되어 안전성과 효과성이 입증된 제품의 경우 간단한 신고절차만 거쳐서 미국으로 수출가능한 제도입니다.
Registration의 경우 매년(미국 회계 기준은 매년 10월임) 정기적으로 Up Date 를 하여야 Active 상태로 유지됩니다.
해당 의료기기가 501(k) 대상인지 Registration 대상인지는 FDA 홈페이지에서 제품에 대한 등급분류를 찾고 등급에 대한 등록 절차를 찾으면 됩니다.